

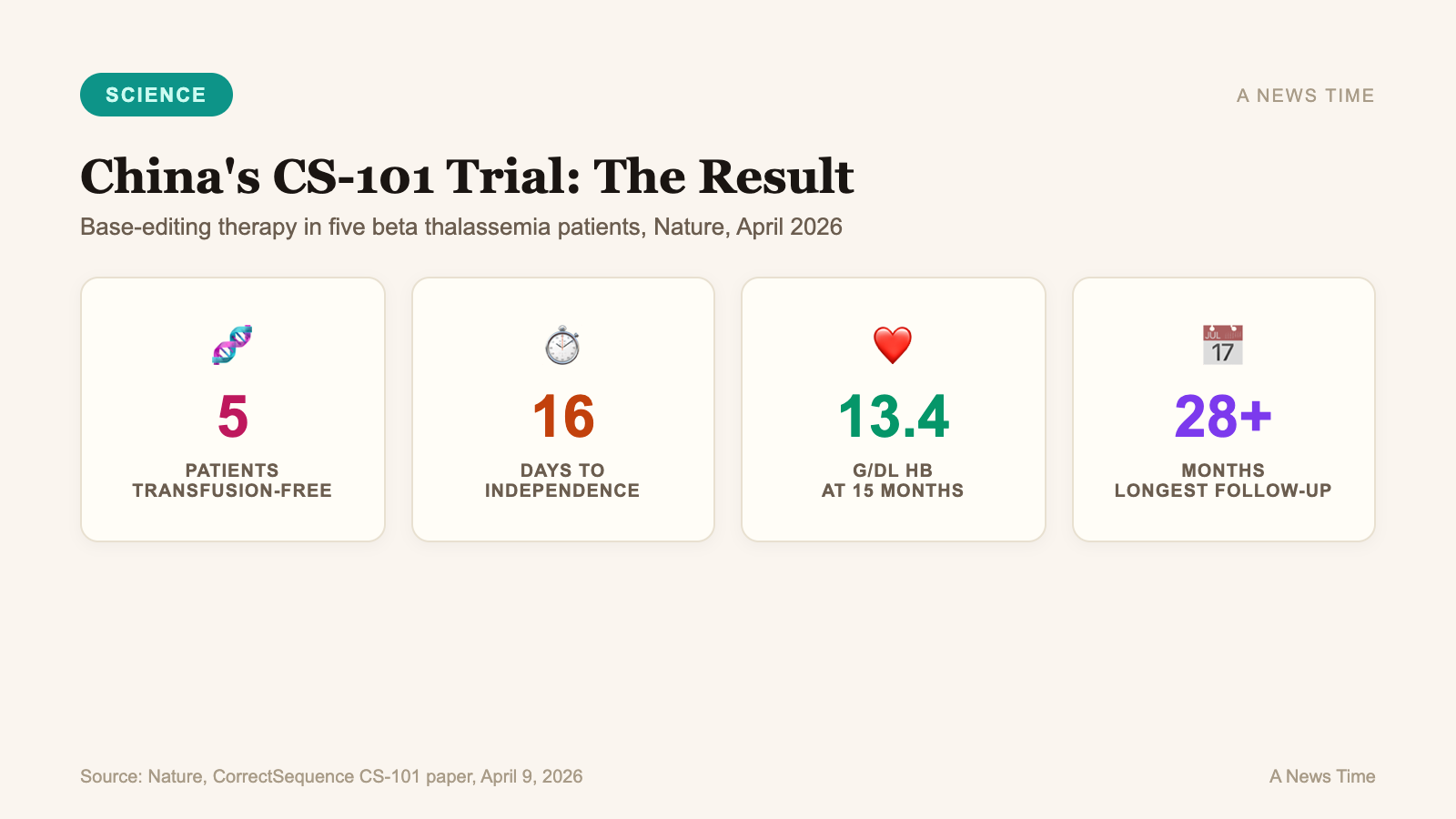
On , the academic journal Nature published results from a small Chinese clinical trial showing that five patients with transfusion-dependent beta thalassemia no longer need regular blood transfusions after receiving a single dose of an experimental gene-editing therapy called CS-101. The trial was run jointly by ShanghaiTech University, the First Affiliated Hospital of Guangxi Medical University, Fudan University, and the Shanghai-based biotech CorrectSequence Therapeutics, which developed the underlying base-editing platform. It is the first base-editing clinical study ever published in Nature, and the first credible signal that gene therapy for inherited blood disease is now a genuinely global field rather than a U.S. and European one.
The five patients in the investigator-initiated trial all achieved neutrophil and platelet engraftment after the edit, and became transfusion-independent an average of just 16 days after infusion. Hemoglobin levels climbed into the near-normal range of 12.4 grams per deciliter within three months and had stabilized around 13.4 g/dL after 15 months of follow-up. As of the November 17, 2025 data cutoff reported in the study, all five patients had maintained transfusion independence for more than a year, with the longest follow-up exceeding 28 months.
What beta thalassemia actually is
Beta thalassemia is a hereditary blood disorder caused by mutations in the beta-globin gene, which encodes one of the two protein subunits that make up adult hemoglobin. When the beta-globin gene is damaged, the body cannot produce functional hemoglobin in adequate amounts, and red blood cells die early. The most severe form, transfusion-dependent thalassemia (TDT), means patients need a blood transfusion every two to five weeks for their entire lives.
Scale helps. The human body contains roughly 25 trillion red blood cells at any given moment, and a healthy adult replaces about 1 percent of them every day. In a child with transfusion-dependent beta thalassemia, that replacement pipeline is broken at the source. Transfusions keep the patient alive, but they introduce iron overload, which damages the heart, liver, and endocrine organs. Until recently, the only potentially curative option was allogeneic stem cell transplantation from a matched donor, which is limited by donor availability and rejection risk.
The World Health Organization estimates that tens of thousands of children are born with transfusion-dependent thalassemia each year, concentrated in Mediterranean, Middle Eastern, South Asian, and Southeast Asian populations where carrier frequency is highest. For those patients, a genuine one-time treatment is the difference between being tethered to a transfusion center and not.
How the edit works
CS-101 is not a conventional CRISPR-Cas9 therapy. Cas9, the enzyme most people associate with CRISPR, cuts both strands of DNA at a targeted site and relies on the cell's repair machinery to make the intended change. That approach works, and it is the mechanism behind Casgevy, the first FDA-approved CRISPR therapy. It is also a blunt instrument in some contexts, because double-strand breaks can cause large chromosomal deletions, rearrangements, or off-target edits that are difficult to screen away.
CorrectSequence's approach is base editing, a newer technique that does not cut both strands of DNA. The company's "transformer Base Editor," or tBE, uses a modified CRISPR complex to chemically convert one DNA base into another at a precisely targeted location. In CS-101's case, the tBE converts cytosine to a base that pairs like thymidine within a narrow sequence window, introducing a small, controlled change rather than a cut.
The edit does not repair the broken beta-globin gene. Instead, it reactivates fetal hemoglobin (HbF), the oxygen-carrying protein used by fetuses in the womb. HbF binds oxygen more tightly than adult hemoglobin and is normally switched off shortly after birth. In beta thalassemia patients, the adult form is broken, so CS-101 disables the HbF off-switch by editing a regulatory region that a silencing protein binds to, allowing fetal hemoglobin to carry oxygen in place of the missing adult protein.
The editing is done outside the body, on the patient's own hematopoietic stem cells. After editing and quality-control screening, patients receive a conditioning chemotherapy, and the edited cells are transplanted back. Within weeks, they reconstitute the blood system and produce enough fetal hemoglobin to eliminate transfusion dependence.
Why the "no double-strand break" detail matters
The technical story most medical readers want to hear is about safety. The research team reported that CS-101 produced no large genomic deletions, no chromosomal rearrangements, and no significant off-target edits in the treated cells that were transplanted back into patients. That is a meaningful contrast with some earlier CRISPR-Cas9 approaches, where large deletions around the cut site have been documented in a subset of edited cells and have been a persistent source of regulatory concern.
"Compared with existing CRISPR-based therapies, CS-101 demonstrated faster activation of fetal hemoglobin, more rapid hematopoietic recovery, and earlier restoration of normal hemoglobin levels. These advantages translate into shorter hospital stays and reduced healthcare burden."
ShanghaiTech University research team, summary of findings published in Nature (April 9, 2026)
John Timmer of Ars Technica, who covered the Nature paper in detail, wrote that the tBE system tethers the base editor to a repair-inhibiting bacterial protein on the guide RNA complex, so mutations only accumulate where all three components have sat on the DNA long enough. The trade-off is efficiency: about 30 percent of targeted cells are successfully edited, roughly half the rate of some competing base editors. The research team argues the safety gain justifies the trade.
Price is the part that could matter most
The United States already has an FDA-approved CRISPR therapy for the related blood disorder sickle cell disease: Casgevy, developed by Vertex Pharmaceuticals and CRISPR Therapeutics and approved in December 2023. Casgevy is a real treatment with real clinical benefit. It is also priced at approximately $2.2 million per patient, making it one of the most expensive medicines ever launched in any category. A second therapy for beta thalassemia, bluebird bio's Zynteglo, is priced around $2.8 million.
| Therapy | Developer | Target disease | Editing approach | Approximate list price (US) |
|---|---|---|---|---|
| Casgevy | Vertex / CRISPR Therapeutics | Sickle cell, beta thalassemia | CRISPR-Cas9, BCL11A disruption | ~$2.2 million |
| Zynteglo | bluebird bio | Beta thalassemia | Lentiviral gene addition | ~$2.8 million |
| CS-101 (experimental) | CorrectSequence Therapeutics | Beta thalassemia, sickle cell | Transformer base editor (tBE) | Not yet disclosed; expected to be significantly lower |
CorrectSequence has not yet disclosed a price for CS-101, and no price will be meaningful until the therapy moves through regulatory approval in at least one market. The pricing context, though, is what makes the competition question real. A Chinese-developed therapy that matches or beats the efficacy of a $2.2 million U.S. treatment, and that is manufactured within a different cost structure, is a different kind of market event than a scientific publication alone. Endpoints News reported that the CS-101 results suggest American drugmakers will face genuine international competition in the gene therapy space for the first time, and that the cost gap is one of the central reasons.
For adjacent context on how the field is wrestling with the cost of one-time curative treatments, see ANewsTime's coverage of Rocket Pharmaceuticals' Kresladi approval for LAD and unconventional drug manufacturing platforms.

What we still don't know
Five patients is a small number. The trial is described in the paper as an early-stage, investigator-initiated safety study, and the conclusions a reader can responsibly draw from it are narrower than the headlines suggest. Evidence supports that the therapy worked in every patient who received it during the follow-up window reported. Evidence does not yet establish how the therapy will perform across thousands of patients with different genetic backgrounds, different thalassemia mutation patterns, and different baseline organ damage from years of transfusions.
Long-term durability is the second open question. Hematopoietic stem cell edits are, in principle, permanent, because the edited stem cells self-renew and pass the edit to their daughter cells. In practice, long-term durability has to be demonstrated rather than assumed, and the field has seen cases where the clonal population of edited cells shifts over time in ways that affect therapeutic output. Twenty-eight months is a promising follow-up window. It is not a 10-year follow-up.
The regulatory path is the third uncertainty. CS-101 has completed Phase I studies in China and has reportedly been used in nearly 20 patients across Chinese and international trial sites. Full Chinese approval would make CS-101 available inside China's market, but U.S. and European availability would require separate filings, local clinical data, and manufacturing inspections. None of that is trivial, and none of it is fast.
What to watch next
Three things will shape whether CS-101 becomes a globally available treatment or remains a nationally significant milestone. First, the confirmatory trial that CorrectSequence runs next: a 20-to-30 patient expansion with at least two years of follow-up would meaningfully raise confidence in the safety profile. Second, the progress of parallel international sites, which would generate data for filings outside China. Third, the response from Vertex, CRISPR Therapeutics, and bluebird bio, the incumbents who now have a credible reason to reassess their pricing and development strategies.
For the five patients in the trial, the experiment is no longer abstract. They are not on the transfusion schedule that defined their lives, and they have not been for more than a year. Whether that result generalizes to the millions of people who share their diagnosis is the question the next stage of research is supposed to answer. On the evidence published this week, it is a question genuinely worth asking.
Frequently Asked Questions
What is beta thalassemia?
Beta thalassemia is a hereditary blood disorder caused by mutations in the beta-globin gene, which results in insufficient production of functional hemoglobin. The most severe form, transfusion-dependent thalassemia, requires patients to receive blood transfusions every two to five weeks for life.
How is CS-101 different from Casgevy?
Casgevy uses CRISPR-Cas9, which cuts both strands of DNA. CS-101 uses a base-editing system called the transformer Base Editor (tBE), which chemically converts one DNA base to another without cutting both strands. The research team reports that this approach produced no detected off-target edits or large chromosomal deletions in the treated cells.
Is CS-101 available in the United States?
No. CS-101 has completed Phase I studies in China and has been used to treat a small number of patients at Chinese and international trial sites. It has not been approved by the U.S. Food and Drug Administration, and any future U.S. approval would require additional clinical data and a formal filing.
How many patients were in the trial?
The Nature paper reported results from five patients with transfusion-dependent beta thalassemia. All five became transfusion-independent after a single dose, with hemoglobin levels stabilizing in the near-normal range over a follow-up period of more than a year.
Why does the price comparison matter?
Casgevy, the U.S.-approved CRISPR therapy for sickle cell and beta thalassemia, is priced at approximately $2.2 million per patient. A Chinese-developed therapy with comparable efficacy could offer significantly lower pricing, which would change access for patients globally and create direct competition for U.S. developers for the first time in the gene therapy field.
Sources
- A 'cure' for five blood disease patients suggests Chinese genetic medicine can compete globally - Endpoints News
- Landmark trial published in Nature: China's novel base-editing therapy brings hope of cure for thalassemia patients - ShanghaiTech University
- Clinical trial shows gene editing works for beta-thalassaemia, too - Ars Technica
- CS-101 base-editing therapy for transfusion-dependent beta thalassemia - Nature (2026)











